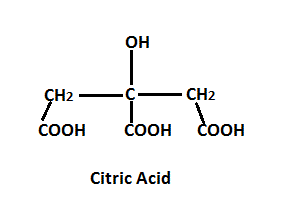
Citric Acid, the well known Krebs cycle intermediate for which the cycle is also known, is normally produced under enzymatic conditions in the mitochondria of cells.
However, the non-enzymatic route to citric acid was unknown until 1977 when Lever Brothers, a division of Unilever, was granted a patent for its chemical
production via reaction of the alkaline earth metal salts, e.g., either calcium isocitrate/alloisocitrate or calcium aconitate at pH's greater than 11.6 and temperatures
of 160°-200°C. This paper will depict the reactions employed and propose the reaction mechanisms using resonance and
The reactions listed herein are actually the reverse of the Krebs cycle and are described in US Patent 4056567 (must be accessed through US patents database).
The starting materials employed for this process can be either the diastereoisomer mixture of isocitrate/alloisocitrate,
the dl mixture of both alone, or cis/trans aconitic acids. Let us begin with the diastereoisomeric mixture (which will also take into account
the
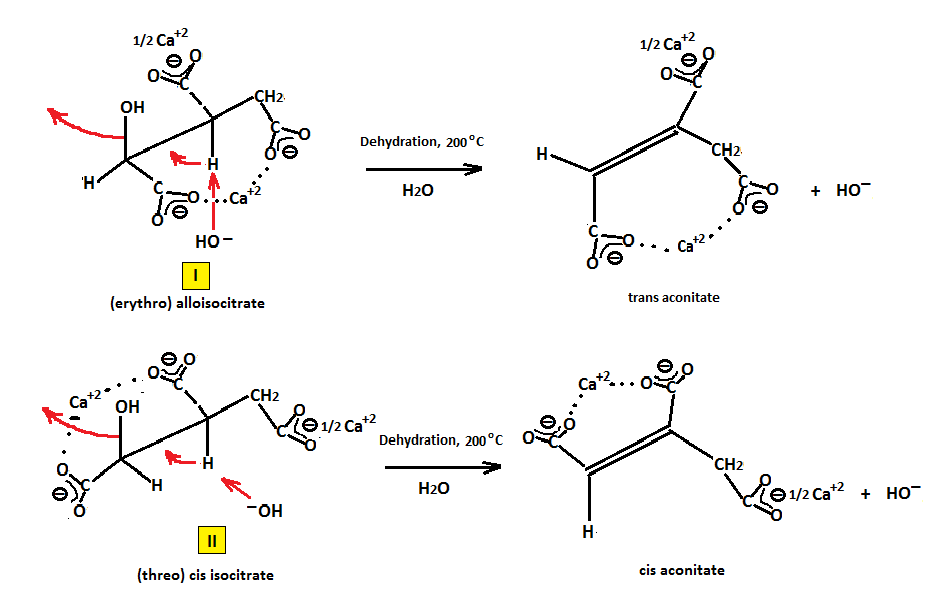
It has been postulated that only isocitrate (I) produces cis aconitate, the presumed reactive intermediate. But since all of the aconitate in the reaction medium has been shown to proceed to citrate it can be further postulated that trans aconitate under the forced conditions of the reaction is isomerized to the cis form either by a low barrier of rotation of the double bond or via some other mechanism.
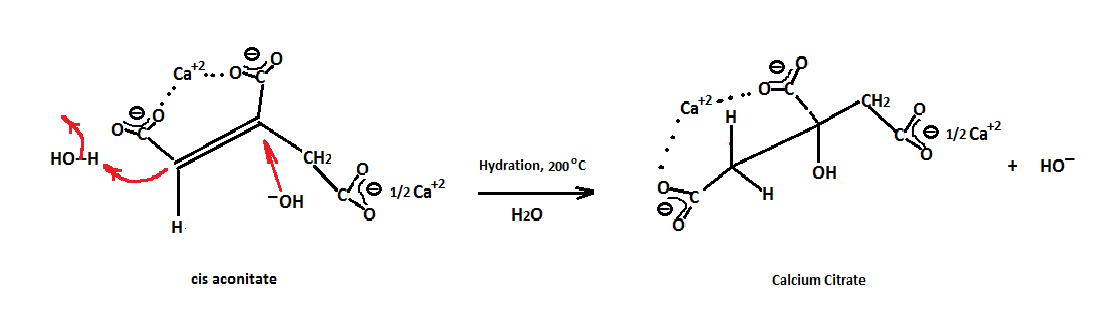
It can also be postulated that conversion may also occur through the partially reactive hydrogens of the CH2 group, which being both "allylic" and α to a carboxyl, is acidic enough to be removed by hydroxide (HO−) ion (step 1.) and placing a negative charge at one of the carbon atoms, followed by abstraction of hydrogen from water (step 2.) and regeneration of hydroxide as shown below. Alkaline earth metal hydroxides especially calcium hydroxide, supply the requisite cation and are basic enough to generate the right basic conditions required for the reactions to proceed. For instance, Ca+2 ions appear to be just the right size and are known for forming stable metal chelates of polycarboxylates. Consequently, in order for the hydration of cis isocitrate to occur, it would be ideal if the two cis carboxylates are chelated and held tightly by Ca+2 on the same side of the double bond forming a very reactive cis intermediate susceptible to a nucleophilic attack by HO−.
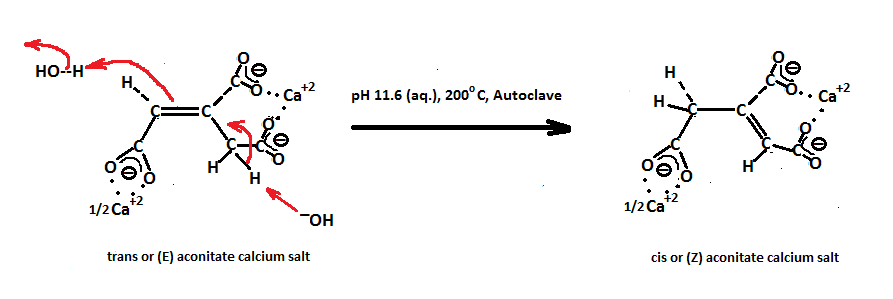
But before we discuss this part, I will depict the resonance forms of cis aconitate and give an insight as to why the the hydroxide (HO−) anion chooses to attack the middle carbon 2 of cis aconitate and not carbon 1.
The resonance structures for cis aconitic are shown below.
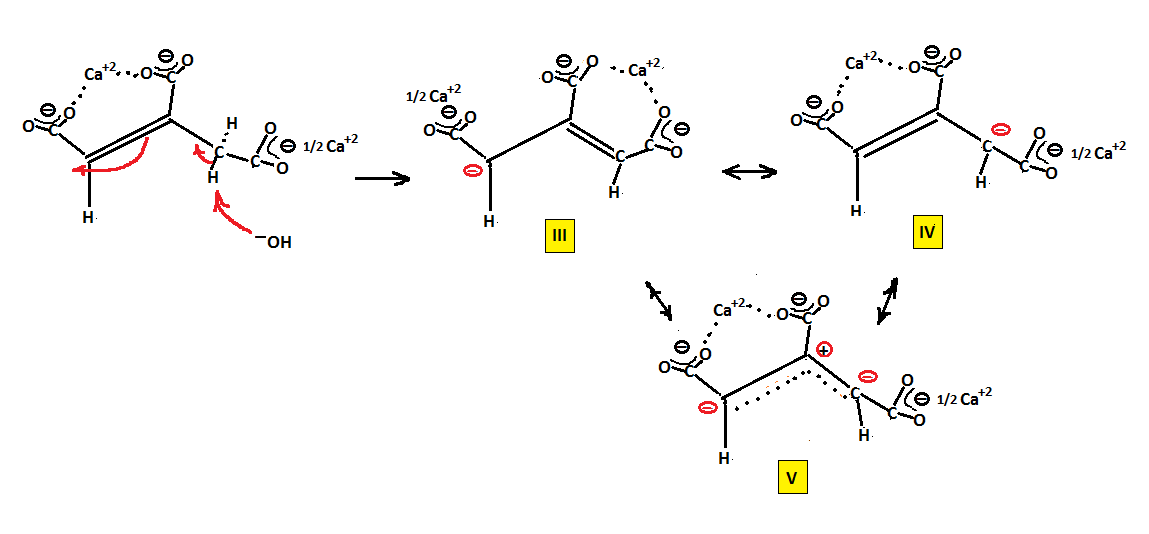
Removing a hydrogen of the CH2 group of cis aconitate via attack of HO− anion and pushing electrons around we can generate structure (III)
which places a partial negative charge − at carbon 3. If we push electrons back we get structure (IV) also having a − charge
and the linear combination of (III) and (IV), resonance hybrid (V). This last structure places a − charge at both the terminal
Although the resonance forms depicts the intermediate structures it still doesn't give us all the information such as how the various groups, e.g., hydroxy, isocitrates, actually interact with one another. Before we proceed to that explanation, let us continue to the actual hydration of cis aconitate to citrate as depicted below.
I mentioned that the reaction takes place at a pH of 11.6. Why this value? Mygel and Sychen (ZH. Khim. 1958 3, 314) have given the value of 11.6 for the pKa4 of the hydroxyl group of citrate. According to the Henderson-Hasselbach equation, if the concentration of the [citrate-4] is equal to that of [citrate-3], then the pH of the reaction mixture should equal to that of the pKa4. The calcium chelated species involving 3 carboxylate groups and one ionized hydroxyl is what probably gives citrate the added stability at this high pH. Normally alkali metal salts at high pHs cause decomposition of citrate under the high temperature and pressure conditions. On the other hand, Silva et al [Biometals(2009) 22:771-778] calculate, via C13 NMR, the pKa4 to be 14.4. However, they state that the substitution at the alpha carbon may modulate the ionization behavior.
Hydration of cis aconitic, therefore, follows the mechanistic scheme below. The HO− anion, acting as a nucleophile attacks electrophilic carbon and pacing a negative charge at carbon 1 (step 1.) and then plucking a hydrogen from water (step 2.) with regeneration of the HO− group. The stability of chelated calcium citrate under the high temperature and high alkaline conditions employed appears to drive the reaction to the right. The stability of the chelated isocitrates at these same conditions are less robust and as shown by the mechanisms above, result in their conversion to the more stable citrate. However, the MO treatment shown below will give a better explanation as to the actual reason these reactions proceed in the manner depicted.
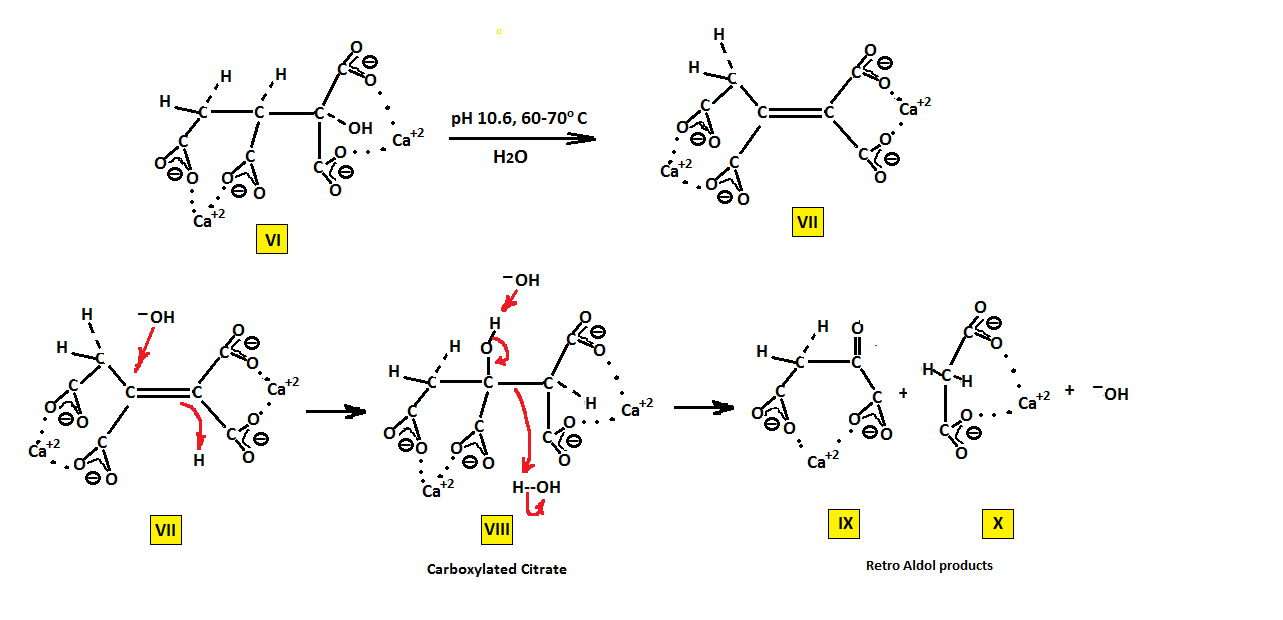
But before this explanation it will be show how a compound similar to what may be considered a hybrid of (E) and (Z) aconitic follows a similar hydration step but produces a highly reactive non isolable product. It will also be shown how increasing the electrophilicity at carbon 2 (middle carbon) can result in an increase in reaction rates.
Compound VII is produced via halogenation followed by saponification of intermediate esters from US patent 4146543, derived from maleic anhydride and US patent 4273935, derived from the monoalkyl ester salt of maleic acid both using calcium hydroxide as base. Under the reaction conditions shown saponification of VI followed by dehydration results in the formation of VII. This carboxylated aconitate being a hybrid of both (E) and (Z) aconitate is shown to hydrate at lower pHs and under atmospheric pressure to produce VIII (NMR analysis shows the disappearance of the CH2 group with time). However, VIII is not isolable and appears to decompose via the retro Aldol reaction into aceto acetate (IX) and malonate (X) both of which exchange hydrogen for deuterium during NMR analysis. Although the intermediate VIII unlike the parent citrate is not a stable entity, its possible formation, as is the case for citrate, points to a similar mechanism.
Aconitate can be visualized as an allylic group with multiple attachments. The CH2 groups occupy a special position on the molecule. They are both α to a carboxylic group and as mentioned allylic. Under certain conditions it should be possible to remove one of these hydrogens and generate an allylic anion a well known 4 π electron (∊) system. An MO description of this allylic anion shows that there are three π orbitals, viz., π1, π2 and π3 as depicted below:
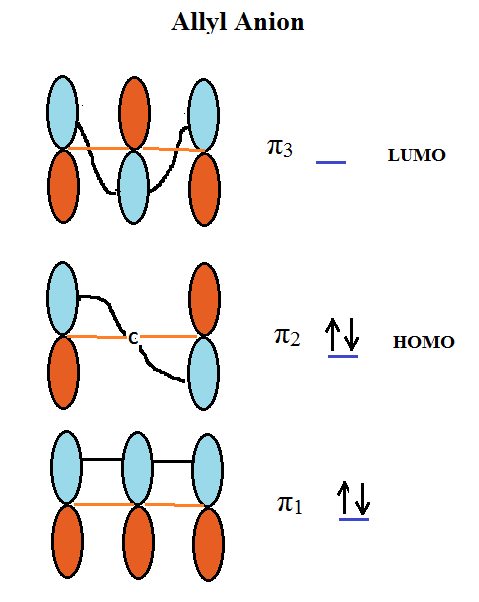
π1 is a bonding orbital with 2∊, π2 is a nonbonding orbital with 2∊ and π3 is an antibonding orbital with 0∊. π2 shows that the wave function is 0 at carbon 2 since a single node passes through it. Because of this the charges in the molecule are mainly at the terminal carbons and not in the middle. This will be shown to be very important for the stability of citrate and also very important for the instabilities of erythro and threo isocitrate under the basic conditions of the reaction.
We begin by postulating that under basic conditions aconitate can be considered a 4 π allyl anion system as the resonance forms shown above. We can depict the interaction of the HOMO and LUMO molecular orbitals, π2 and π3, of the trisubstituted allyl anion of aconitate in the manner shown below where it can be seen that the HOMO of the filled π2 of the allyl can interact with the empty σ* orbital of a hydrogen on water. As stated previously, since the wave function at the central carbon is zero, i.e., a single node passes through it, no interaction at the central carbon occurs.
We can start by saying that the HOMO of hydroxide anion interacts with the LUMO of the empty π3 orbital of the allyl group at the central lobe but not at any of the π3 terminal lobes. Since the empty σ* orbital on hydrogen can only interact with the terminal lobes the only option for the HOMO of the hydroxyanion is the middle lobe of π3 and its interaction with the two terminal lobes of π3 is strictly forbidden. Therefore, we can unequivocally state that only the interactions on the left hand side are allowed.
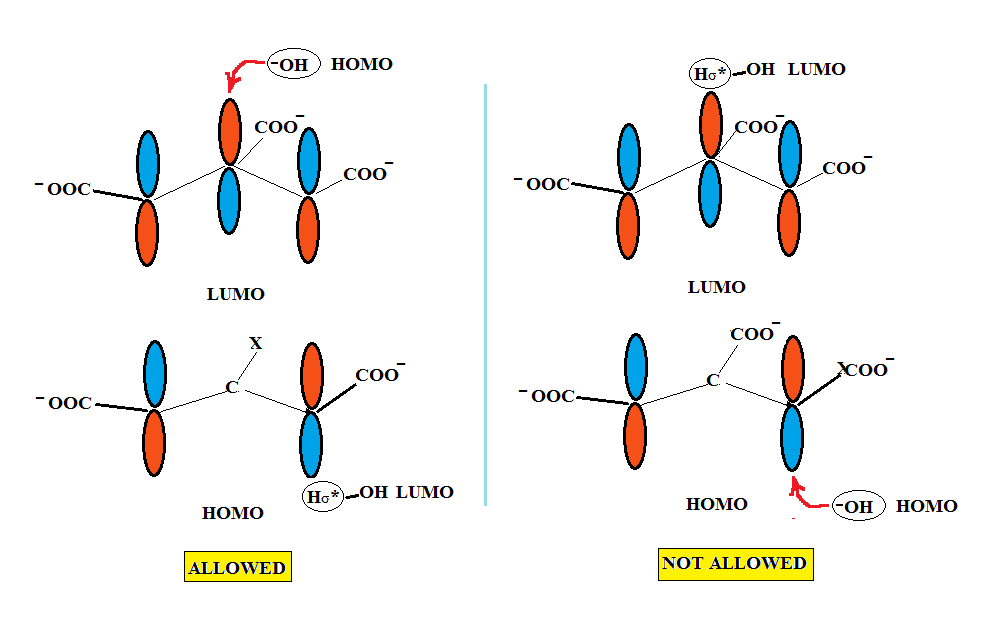
If the mode of addition of water to aconitate were to occur as shown on the right hand side, non allowed HOMO-HOMO and LUMO-LUMO interactions would occur furnishing only isocitric/alloisocitric. However, under the reaction conditions only citric acid is the final product. Generation of isocitric/alloisocitric via this route is not feasible because of the instability of these compounds to the conditions employed. Consequently, I propose that the HOMO-HOMO and LUMO-LUMO interactions appear to be the cause of this instability and is the major driving force for this instability.
Part II is the second paper in the series and will discuss what I consider a theoretical route to the erythro and threo isocitrates via hydration of (E) and (Z) aconitates and will show how the HOMO-LUMO interactions on a substituted ethene group would give rise to isocitrates.
Go back to homepage.
Copyright © April 2015 (revised 5/26/2018) by Eddie N Gutierrez E-mail: edguti144@outlook.com